<-Home
Human Paralogy Server
 OVERVIEW
OVERVIEW
The long-term goal of our laboratory is to understand the evolution, pathology
and mechanism(s) of recent gene duplication and DNA transposition within the
human genome. Our research specifically addresses a new paradigm that has
emerged in the past few years regarding the dynamic nature of human genome
structure. Particular chromosomal regions have been shown to be active in the
acquisition, duplication and dispersal of large gene-containing genomic
segments. We hypothesize that these “jumping genomic segments” are part of
an ongoing evolutionary process that results in a novel form of large-scale
variation in human genomic DNA and contributes rapidly to primate gene
evolution. The large blocks of sequence similarity generated by this process, we
further propose, provide the substrates for aberrant recombination, thereby
leading to recurrent and potentially pathogenic chromosomal structural
rearrangements.
The general aims of our research are:
- to investigate the molecular mechanism(s) responsible for recent
duplications through comparative sequencing and phylogenetic reconstruction
of duplication events.
- to evaluate the role the duplications play in both primate chromosomal
evolution as well as the emergence of new genes and gene families
- to develop computational methods to rapidly identify and characterize
highly homologous duplications in man and other vertebrates
- to assess the contribution of duplication architecture to genomic
instability associated with genetic (genomic) disease.
SPECIFIC AREAS OF RESEARCH
Pericentromeric Organization
Genome-Wide Analysis
Rapidly Evolving Human Genes
Experimental and Computational Methods
I. The Dynamic Structure of Human Pericentromeric DNA
We have identified that the pericentromeric regions of specific human
chromosomes have been hotspots for recent genomic transposition and duplication.
The available data indicate that genomic segments ranging in length from 5-150
kb have been directed to the pericentromeric DNA, that specific repeat sequences
serve as preferred sites for integrations and that these segments transposed to
these regions very recently (1-15 mya) through a complex series of events.
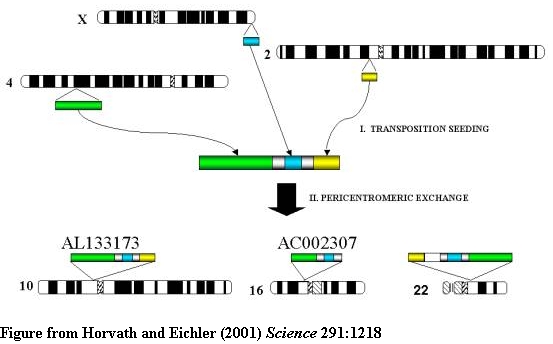
TWO STEP MODEL OF PERICENTROMERIC DUPLICATION
A two-step model for the origin and dispersal of recently duplicated segments in
the human genome. Genomic segments of various lengths from different regions of
the genome were duplicated to an ancestral pericentromeric region followed by
the dispersal of a mosaic genomic segment to multiple pericentromeric regions.
Green, 85 kb from 4q24; blue, 9.7 kb from Xq28; yellow, 10 kb from 2p12.
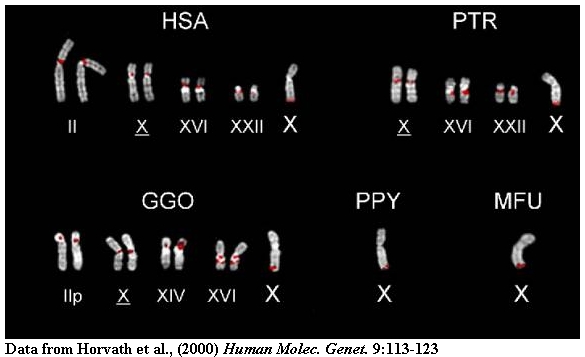
We have documented considerable variation in the genomic architecture of these
regions among closely related primates. In addition, these studies have shed
insight into the structure and organization of euchromatic/heterochromatic
transition regions of the human genome.
The duplications within pericentromeric regions are structured as duplications
within duplications. Individual modules from diverse regions of the genome are
juxtaposed to form larger cassettes that subsequently duplicated between
non-homologous chromosomes. The underlying mechanism for these secondary events
is unknown although breakpoint analysis does suggest the involvement of
satellite sequence.

Based on our genome wide-analysis, two different complex models for their
organization have been proposed: endoduplication of genic cassettes 19p12 and
large-scale inter/intrachromosomal transposition (15q11 and 16p11). The latter
organization contributes to recurrent chromosomal structural rearrangement
associated with human genetic disease. We are committed to the further
characterization of these complex regions of the genome and the development of
assays to correlate their dynamic structure with chromosome function, gene
evolution and human disease.
II. Paralogous Structure of the Human Genome
As part of the International Human Sequencing Consortium, we examined the
distribution of highly homologous (90-98% sequence identity and >1 kb in
length) duplications throughout the genome and the quality of sequence assembly
within exceptional chromosomal regions. Our analysis revealed that a large
fraction of the genome (5-10%) consists of large duplicated segments often
containing pieces or entire genes. The amount of duplication is more than most
scientists would have anticipated. 5-10% translates into about 2 chromosome's
worth of DNA. What is more surprising than the amount, however, is the way this
material is distributed. Most scientists believed that highly homologous
duplications would be restricted to clusters (tandem arrays of genes). What we
have found is that these pieces have been mobilized (akin to transposons) to
various regions of the human genome. These same regions are known to be unstable
and associated with disease. This mosaic nature suggests that the genome is much
more plastic than anticipated and has important implications for protein domain
accretion during vertebrate chromosomal evolution.

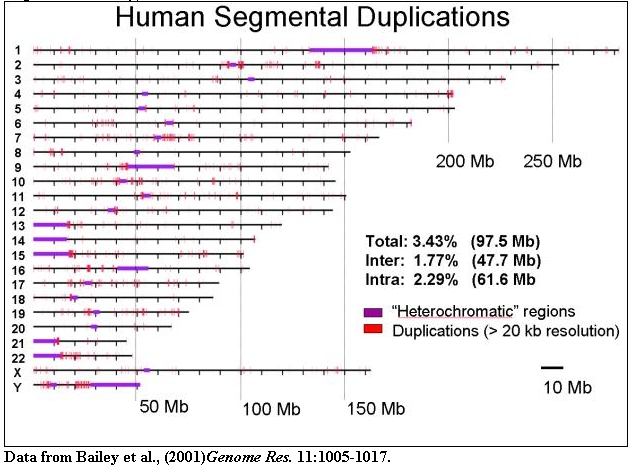
The organization of segmental duplications within human chromosomes is shown
based on the October assembly of the human genome. Duplications (>20 kb in
size and >90% sequence identity) are indicated.
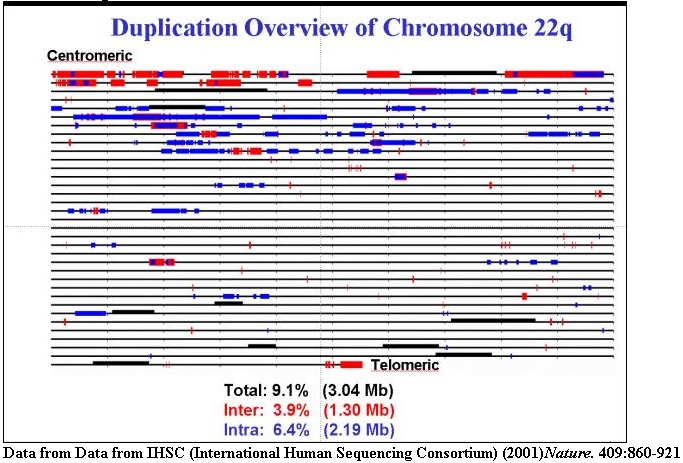
An expanded view of recent segmental duplications on human chromosome 22 (1% of
the genome). Inter (red) and intrachromosomal (blue) duplications are shown.
Many of these regions are sites of recurrent chromosomal structural
rearrangement associated with human genetic disease.
III. Positive Selection of Novel Hominoid Genes
The recent duplication and transposition of a 20 kb duplication throughout
hominoid chromosome 16 (HSA16). The duplication contains an uncharacterized gene
family, termed morpheus, which emerged ~7 mya and subsequently spread throughout
the chromosomes of man and the great apes.
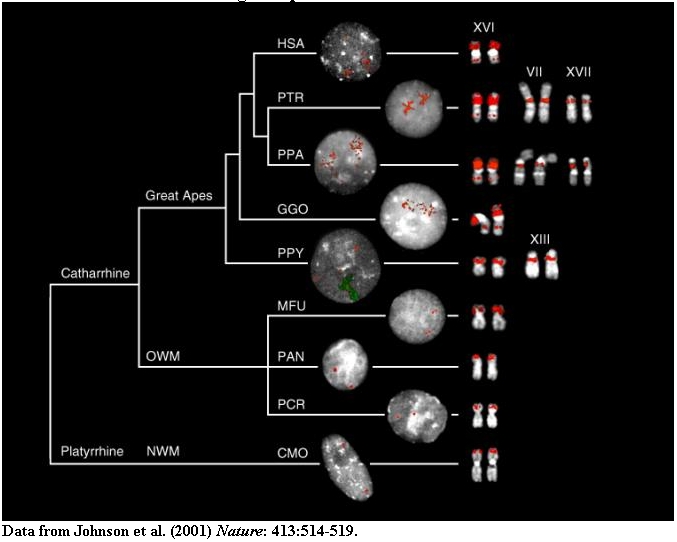
Metaphase (right) and interphase nuclei (left) from a representative panel of
Old World (OWM), New World monkeys (NWM) (MFU=Macaca fascicularis, PCR=Presbytis
cristata, PAN=Papio anubis and CMO=Callicebus mollochus) and hominoid species
(HSA=Homo sapiens, PTR=Pan troglodytes, PPA=Pan paniscus,
GGO=Gorilla gorilla, PPY=Pongo pygmaeus) are shown that have been hybridized with probes (16.1/9 and 16.8/12).
The results are depicted in the context of a generally accepted phylogeny of the
species (Ref. 6). Roman numerals above metaphase chromosomes are according to
standard cytogenetic nomenclature. Note the multiple copies of the repeat
located on XVI among the hominoids which effectively appear to paint the short
arm of the chromosome. Reciprocal experiments using probes derived from other
primate species were used to eliminate the possibility of false negative signal
(Methods). The orangutan interphase also shows hybridization of a human
chromosome XVI paint (green fluorescence). In this species, copies of the LCR16a
duplicon have spread to the pericentromeric region of chromosome XIII.
The rapid evolution of protein-coding exon 2 (right) of the morpheus gene family
as compared to the intronic regions (left). The data suggest that the exons have
evolved much more rapidly than the intronic regions. >95% of the changes have
resulted in amino-acid replacements among paralogues indicative of positive
selection. The function of this gene family is being explored.
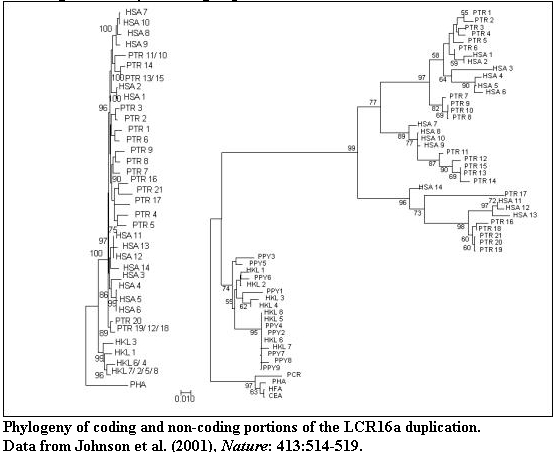
Neighbour-joining phylogenetic trees for a) 1421 bp of intronic sequence (introns
2,3,4, Fig. 1c) and b) 186 bp of exon 2 are compared. Extreme positive selection
for exon 2 is indicated on the branch separating humans and African apes from
the orangutan lineage (a 35-fold excess of amino-acid changes when compared to
the neutral model). Note the significantly shorter branch lengths for flanking
non-coding intronic sequences which are consistent with nucleotide sites
evolving at a neutral rate. More than 95% of the informative sites for the
phylogenetic tree of exon 2 are the result of amino-acid altering nucleotide
changes. Branches showing significant positive selection are indicated by arrows
with accompanying ba/bs ratios (estimated amino-acid replacement and synonymous
changes per branch per site, Ref. 12). Significance was calculated based on the
difference (*p<0.05, **p<0.01). Sequence for various duplicate copies are
identified by species acronym (PHA=Papio hamadryas, HKL= Hylobates klossi and
see Fig. 2) and a number corresponding to clone and/or accession within GenBank
(Table 3 Supplement). Scale bar, Jukes-Cantor corrected distance. The midpoint
of all trees was set to 1/2 the distance between gibbon and baboon sequence taxa.
Only bootstrap values >50% are shown (n=1000 replicates). A similar topology
showing positive selection for exon 4 sequence was obtained (Supplement Fig. 3).
IV. Characterization of Low-Copy Repeats within the Human Genome
PCR amplification from monochromosomal source material provides evidence of interchromosomal duplications.
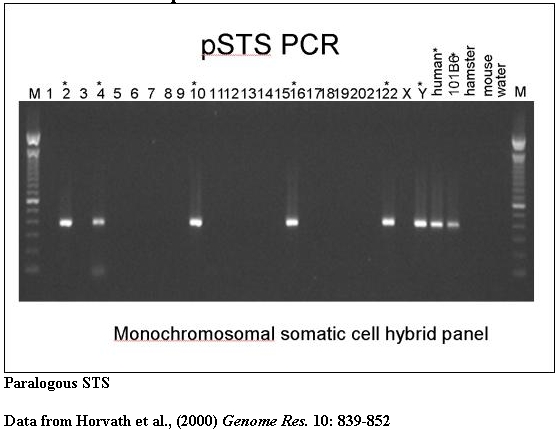
A typical PCR amplification of a paralogous STS against a panel of monochromosomal somatic cell hybrid DNAs. pSTS1 was designed to 101B6 (chromosome 2) sequence yet amplified a ~383bp product from chromosomes 2, 4, 10, 16, 22 and Y (marked with asterisks).
Sequencing of PCR products provides diagnostic sequencing variants that may then be used to recover BACs from existing libraries for large-scale sequencing. Such BACs are submitted to sequencing centers to ensure that these highly duplicated regions are included in the final assembly of the human genome.

The PCR products from pSTS 1 were bidirectionally sequenced and aligned (Consed). Basepairs in bold represent 101B6 basepairs while the numbers above each bp represent its location in 101B6. Only the paralogous sequence variants (PSVs) which distinguish each chromosome are shown a period represents the same bp as 101B6. Along the right are the sequences of the monochromosomal hybrid sequence (MCH). Below each chromosomal sequence signature, a subset of RPCI-11 BAC clones corresponding to each PSV is indicated. The numbers correspond to pSTSs developed to the 101B6 reference sequence. Similar analyses were performed for 17 other pSTS.
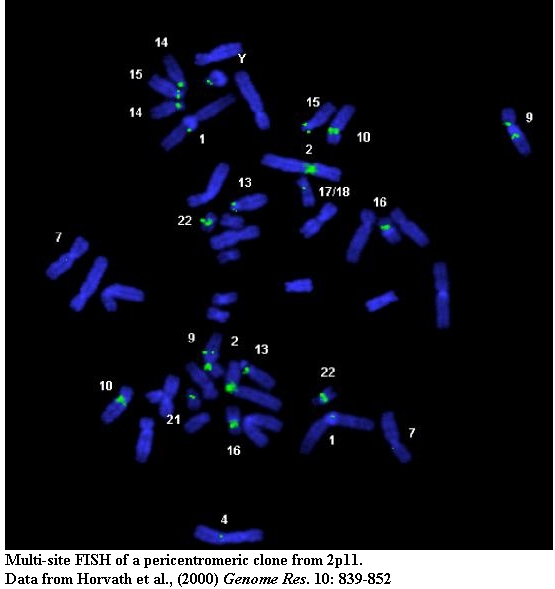
Standard cytogenetic techniques (FISH and extended chromatin analysis) help to characterize patterns of inter and intrachromosomal duplication.
Sequence-based and cytogenetic methods are combined with in silico analysis to provide a global overview of duplications. Much of the computational work in the laboratory is performed using a 32-node LINUX cluster farm. Specific software for large-scale sequence manipulation and duplication analysis has been developed to analyze datasets from entire genomes.

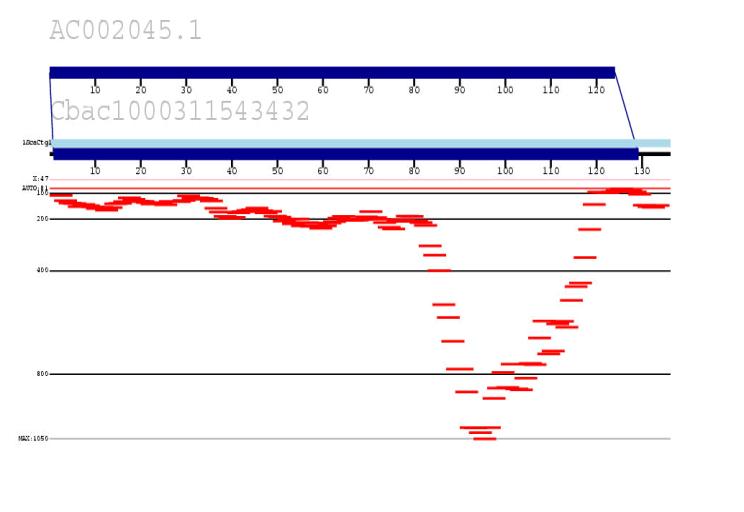
We are developing an in silico method to identify all recent highly homologous duplications within the human genome by combining public and private (Celera) data. The approach measures the depth of coverage of random read data (Celera) against human reference sequence (public accessions). Statistically significant pileups of reads (when compared to known unique sequence) are indicative of duplicated regions (shown in red). Our goal is to use this data to generate a true paralogy map of the human genome, identify sequence gaps and begin to characterize regions of genomic instability associated with disease and human polymorphism.