Jump To:
Overview
Genomic duplication followed by adaptive mutation is considered one of the primary forces for the emergence of new genes in a species. Duplicated sequences are also a major driver of genetic structural variation because of their potential to mediate unequal crossing over (Fig. 1). The long-term goal of my research is to understand the evolution, pathology, and mechanisms(s) underlying recent segmental duplication. Our work involves the systematic discovery of these regions, the development of methods to assess their variation, the detection of signatures of rapid gene evolution, and ultimately the correlation of this genetic variation with phenotypic differences within and between species.
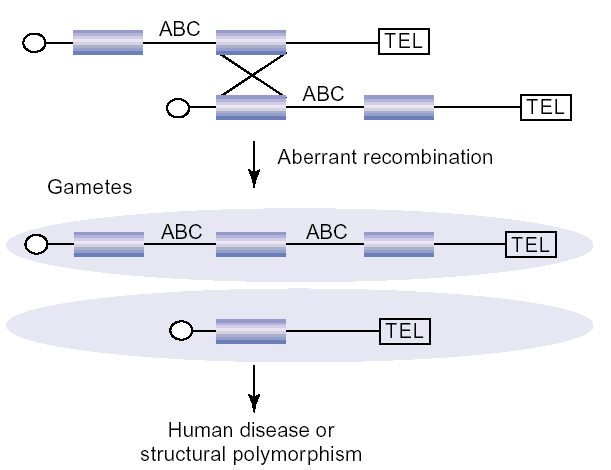
Non-allelic homologous recombination (NAHR) between blocks of segmental duplication (blue) during meiosis leads to microdeletion and microduplication of the unique region bracketed by duplications. If the region contains dosage-sensitive genes (ABC), disease may result. If not, the duplicated chromosome is predisposed to additional rounds of microdeletion and duplication with increased probability.
Summary
Our research program has focused on understanding the functional and structural impacts of segmental duplications on the human genome. Duplications may be viewed as dynamic mutations (Fig. 1)—an initial event increases the probability of a second mutational event. Sequence homology created as a result of duplication increases the probability of additional rounds of gene conversion, unequal crossing-over, and subsequent rearrangement. Not surprisingly, many of the largest duplication blocks are substrates for recurrent chromosomal structural rearrangements associated with human developmental disorders and disease susceptibility. These particular regions of the genome represent hotspots of evolutionary and contemporary change. Perhaps paradoxically, these regions are also rich in predicted protein-coding genes, yet these genes have been historically understudied and poorly annotated because of their inherent repetitive nature. We investigate their impact of human disease and evolution.
Our research has challenged the field of vertebrate genome evolution, which largely held that “most of nature’s experiments with duplication must have been done at the stages of fish and amphibians.” Within the genetics and evolutionary community, this has led to a resurgence of interest in genomic duplications and a series of unanswered questions: How did this complex architecture of duplications evolve in humans? What is the underlying mechanism? How variable are these regions within the human population and to what extent do they contribute to disease and phenotypic differences? How does the human structure compare to that of great apes and other mammals? Have new genes evolved by this mechanism that are important for human/great ape adaptation? Our research program is committed to addressing these questions.
Structural Variation and Disease
From our research, we identified 130 regions of the human genome that are prone to large-scale chromosomal rearrangement based on the architecture of segmental duplications. We used this duplication road-map to identify dozens of recurrent deletions, duplications, and inversions associated with neurodevelopmental disorders, including autism, developmental delay, and epilepsy. Using long-read sequencing platforms to construct telomere-to-telomere (T2T) genomes, we developed approaches to simultaneously discover and phase all forms of human genetic variation, including single-nucleotide substitutions, insertions/deletions (<50 bp), and structural variants (≥50 bp) in both controls and patients with unexplained genetic disease (Fig. 2). We hypothesize that structural variation (deletions, duplications and inversions) is an underestimated mutational force contributing to genetic disease—particularly disease susceptibility loci. Because the sum total of all mutations in an individual contribute to disease risk, we believe that completely sequenced and phased genomes will become the standard for all clinical genetic tests. We are developing approaches to genotype and associate these more complex forms of genetic variation to account for the missing heritability of human genetic disease.
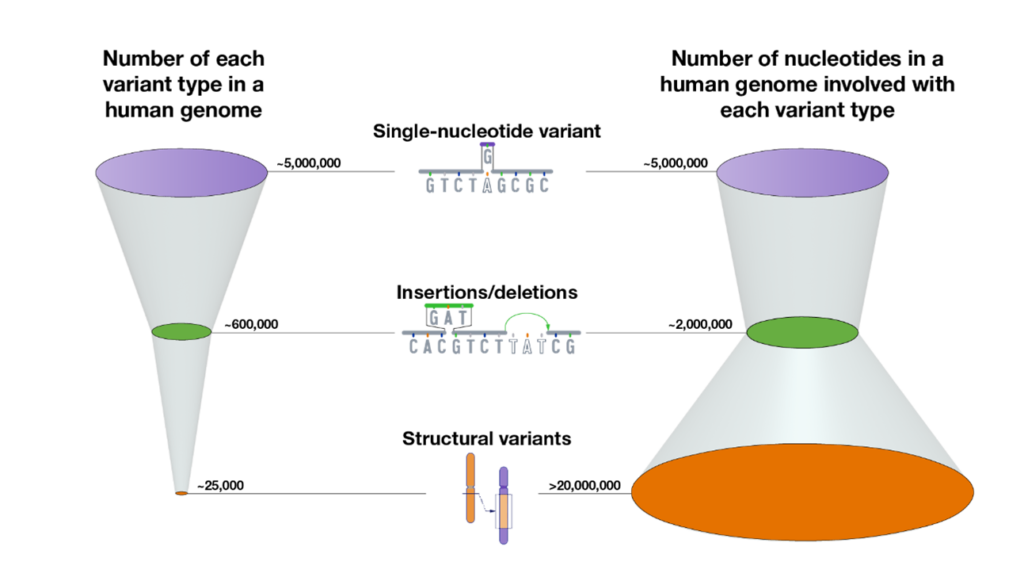
Nearly complete sequencing of genomes telomere-to-telomere has revealed that the average diploid human genome carries between 4-5 million single-nucleotide variants, ~600 thousand insertions/deletions, and ~25 thousand structural variants. While far fewer in number, structural variants affect much more of our genetic code and, thus, are thought to be more consequential. Because of their association with repetitive DNA, however, structural variants have been understudied as part of standard genetic association studies.
Image credit: Darryl Leja, NIH/NHGRI
Ape Genome Evolution
As a complement to our analysis of human variation, we focus on understanding natural genome variation between humans and other primates. We compare the pattern of human segmental duplication to other primate species (chimpanzee, gorilla, orangutan, macaque, marmoset, lemurs, etc.). Each of these species represents a distinct phylogenetic branchpoint from the human lineage and, as such, provides us with temporal snapshots of genome mutation allowing us to distinguish lineage-specific versus ancestrally shared segmental duplications and structural variation. The long-term goal of our research program is to understand the evolutionary history of every base pair of the human genome with a particular focus on regions of segmental duplication. Using long-read sequencing, we recently completely sequenced the genomes of several primate genomes and constructed an evolutionary model for the origin and spread of segmental duplications over the last 25 million years of primate evolution. We find that interspersed segmental duplications arose nonrandomly in time and space, segmental duplications are preferential sites for large-scale rearrangements that distinguish the chromosomes of apes—especially inversions, and that specific sequences that we term “core duplicons” are the focal point for the accumulation of intrachromosomal segmental duplication blocks independently among closely related ape species. We are currently refining the breakpoints of these large-scale genetic changes to understand their mechanism of origin and their potential to impact the formation and regulation of genes (Fig. 3).
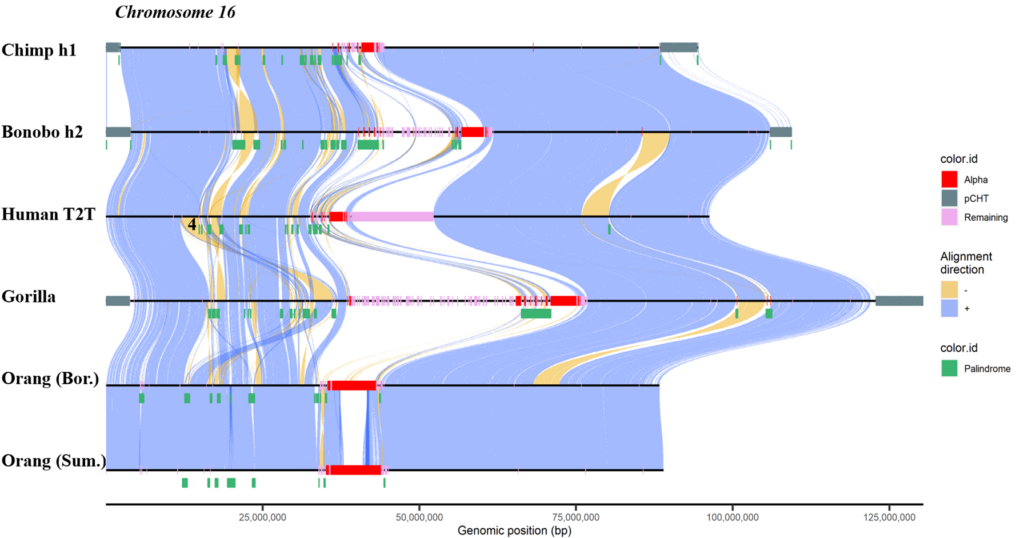
A comparative plot showing the complete sequence of human chromosome 16 to syntenic chromosomes from other apes. Large blocks of synteny (blue) are disrupted by numerous inversions (yellow), palindromic segmental duplications (green), and satellite DNA (red, pink, gray). Adapted from Yoo et al., 2024.
The Emergence of New Protein-coding Genes in Human
The process of segmental duplication provides a vehicle for primate gene innovation in two different ways. First, duplications may lead to the adaptive evolution of genes “liberated” from the selective constraint of ancestral function. Second, the accumulation of diverse duplications at prescribed locations in the genome juxtaposes different gene cassettes in novel genomic texts. This has led to the formation of chimeric genes in a process akin to “exon shuffling.” Although most random mutations create duplicate pseudogenes, occasionally functional products may emerge. One exciting highlight of our research has been the discovery and characterization of rapidly evolving genes and fusion genes specific to the human and great-ape lineages. This includes numerous genes implicated in the expansion of the human frontal cortex (e.g., ARHGAP11B, SRGAP2C, NOTCH2NL, TBC1D3, etc.) as well as gene families that have been poorly characterized but show signatures of positive selection in humans (NPIP, GOLGA6/8, RGPD, etc.). We are using long-read sequencing techniques to characterize full-length transcripts, open-reading frames, and the subset of newly minted genes that are intolerant to mutation and, therefore, more likely to be functional. We challenge the 50-year-old paradigm that the majority of functional differences that distinguish humans from chimpanzees are regulatory in nature; rather, we hypothesize that lineage-specific protein-coding genes arose in each ape lineage as a result of segmental duplication. We are committed to understanding the function of these newly minted genes and their role in human health.
