Research Program
Primate Genome Evolution and the Mechanism of Origin:
As a complement to our analysis of
human variation, we focus on understanding natural genome variation
between humans and other primates. We compare the pattern of human
segmental duplication to other primate species (chimpanzee, gorilla,
orangutan, macaque, marmoset and lemur). Each of these species
represents a distinct phylogenetic branchpoint from the human lineage
and, as such, provides us with temporal snapshots of genome mutation
(Fig. 2). Cognizant of the limitations of assembled genome sequence, we
employ the tools we have developed during the analysis of the human
genome to systematically characterize lineage-specific and shared
duplications. In addition to genome-wide analyses, targeted high
quality sequencing of large insert clones will provide long-range
continuity to model evolutionary processes within these regions.
There are three objectives:
- We reconstruct the evolutionary history of every recent (<40 million years) segmental duplication within the human genome. We are developing methods to unambiguously identify ancestral states based on outgroup genomic data and to extract historical associations by application of A-Bruijn graph theory which promises to deconvolute the subrepeat structure of mosaic duplications. Using comparative sequence from these regions, we are also currently modeling the frequency of gene conversion and its impact on the structure of these regions. Such a comprehensive duplication analysis is unprecedented and will provide us with fundamental insight into rates of fixation, gene conversion and episodes of duplication expansion.
- Our second objective is to understand the underlying mechanism of segmental duplications. We have recently developed a donor-acceptor model for human duplications which indicates that Alu repeats are key elements for the mobilization of duplications, while low-complexity (GC and AT rich) sequences may account for the preferential integration of these elements into specific chromosomal regions. We propose to directly test this model by identifying and characterizing lineage-specific duplications that have inserted into new locations within human, chimpanzees and gorillas genomes. Studying the phylogenetic relationship of such sequences to their antecedents will provide fundamental insight into putative donor and acceptor sequences at the sites of transposition and integration, respectively. To date, we have cloned and mapped 29 of these new insertion sites within gorilla, chimpanzee and orangutan. Large-scale sequence analysis of seven integration sites suggest coordinated deletions of repeat-rich insertion site (5-16 kb) during the segmental duplication process. Ultimately, these data will serve as the basis for future experimental modeling of this process.
- Our third objective is to understand the molecular basis of evolutionary chromosomal rearrangements. Among mammals there is strong correlation between breakpoints in conserved synteny and segmental duplication. It is unknown, however, whether segmental duplications are the cause or consequence of the rearrangement process. We propose to assess the impact of segmental duplications and chromosomal speciation breakpoints by analyzing two primate taxa, gibbon and squirrel monkey, which show accelerated rates of karyotype evolution when compared to other primate and most mammals. Our approach will be to randomly generate paired-end sequence from large-insert BAC libraries from each species and optimally align these sequences to the human reference genome. BAC-end sequences that show discordancy from a collinear order from human will be selected for experimental validation by FISH. Large-insert clones that span breakpoints (translocation, inversion or fusion) will be sequenced effectively allowing us to traverse the site of rearrangement. The high degree of sequence identity (>90%) with human DNA should allow breakpoint positions to be refined to the level of the basepair, which is currently impossible with other mammalian sequencing projects. Based on our preliminary analysis of chimpanzee, we predict that >50% of all breakpoints will associate with segmental duplication. Comparative analyses will be used to determine the timing of the duplication in relation to the rearrangement. We propose that bursts of segmental duplication precipitated lineage-specific accelerations in large-scale chromosomal rearrangement within gibbon and the squirrel monkey lineages.
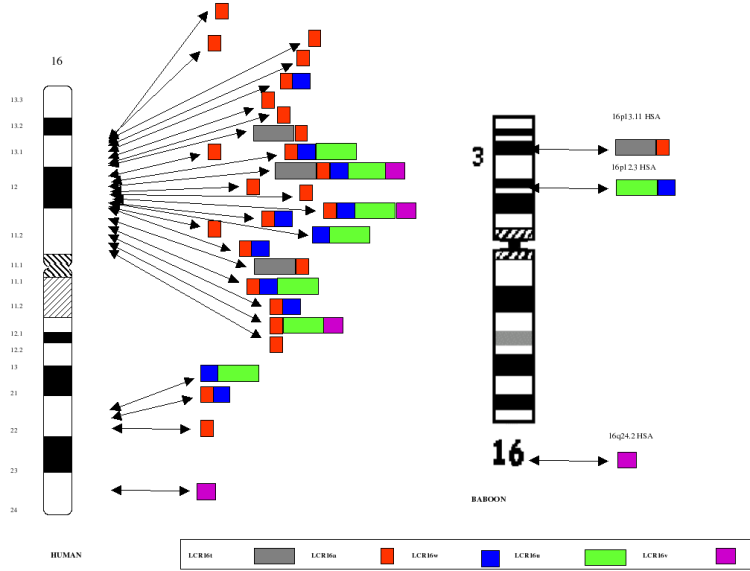
Figure
2: Human vs. Baboon Duplication
Architecture. The organization of five LCR16 (low copy
repeats on chromosome 16) segmental duplications is compared between
human and baboon. In humans, the duplications range in size from
19-75 kb, are 97-99.5% identical and are distributed in different
permutations to 27 different map positions shown along the
ideogram. In baboons (and other Old World monkeys), the
corresponding segments are not duplicated and map to a single
locus. The data suggest a dramatic expansion of segmental
duplications during hominoid evolution on this chromosome. Note: The
LCR16a duplication (red) contains a novel gene family (morpheus) that
shows positive selection only in humans and the great-apes.